
Quantum is Cleaner – Conformational Analysis in Ligand-based Drug Design
Drug discoverers require high throughput technology in both experimental and computational (virtual) library screening workflows to offset the extreme complexity in designing and testing new drug candidates. For the latter, the technological landscape is dominated by informatics and classical physics (mechanics) modelling using empirical force fields (FF). While force field accuracy has steadily improved over the last 50 or so years (e.g. mm3 to OPLS, CHARM) the reliance on FF approaches introduces an alchemical core that may be hindering the analysis and identification of new drug targets.
It is well known and accepted that quantum chemical approaches to molecular property prediction (shape and electrostatics) would add an improved data quality and reliability, but a lack of familiarity with quantum methods has resulted in reluctance to shift to a new quantum regime, exacerbated also by historical limits in required computing power to achieve the same throughput – the speed /accuracy trade-off has been unfavorable.
To address the throughput limitations, ChemAlive has built ConstruQt – a cloud powered high throughput quantum chemistry software and database. With this tech ChemAlive has produced a database of 3.5 million molecules at the semi-empirical quantum chemical level, PM6. ConstruQt, which was designed with high throughput in mind, allows molecular design at comparable throughput (for lead optimization) to more traditional classical methods by taking advantage of automation and massive compute-power scaling on the Cloud.
Following recent literature on the improved results for conformational analysis (shape prediction) and its own intuitive nose, the ChemAlive team has performed analyses of its large database and disclosed a number of unique aspects confirming the jump in accuracy and utility (scope) when using quantum chemistry calculations for understanding molecular structure (see previous blog posts).
Now, the ChemAlive team has partnered with Chemspace. to demonstrate the power of quantum chemistry specifically in drug discovery and ligand-based molecular library screening and design at the lead stage. We now disclose a case study where the ConstruQt software and data has been connected to Chemspace fragment library molecules. The guiding principle of the study is to achieve more accurate ligand-based analytics on potential molecules that are built from readily available reactive fragments purchasable on Chemspace. Thus, the availability and ease of synthesis is encoded in the final library allowing for fast turnaround in lead optimization.
Here is our approach presented stepwise:
- We chose a known pyrazole based drug and identified a substructure from it as an ‘active’ moiety.
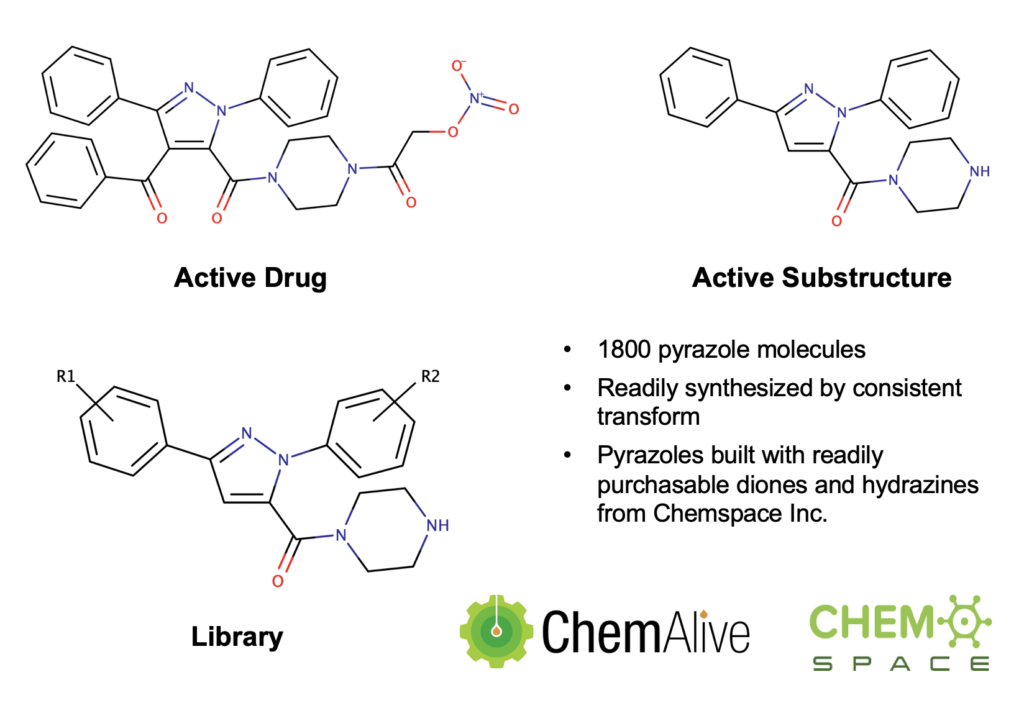
- With Chemspace, we have enumerated 1800 pyrazole molecules based on readily purchasable diones and hydrazines where the molecules contain this substructure from above. This is the Knorr Pyrazole named reaction. We note that the REAXYS database of hydrazines and diones would allow for over 27 million possible pyrazole products using this methodology from known starting materials.
- We have used ConstruQt to compute the conformational space with quantum chemistry (PM6) and force field mechanics (UFF) on these 1800 molecules plus the reference drug molecule in an active pose.
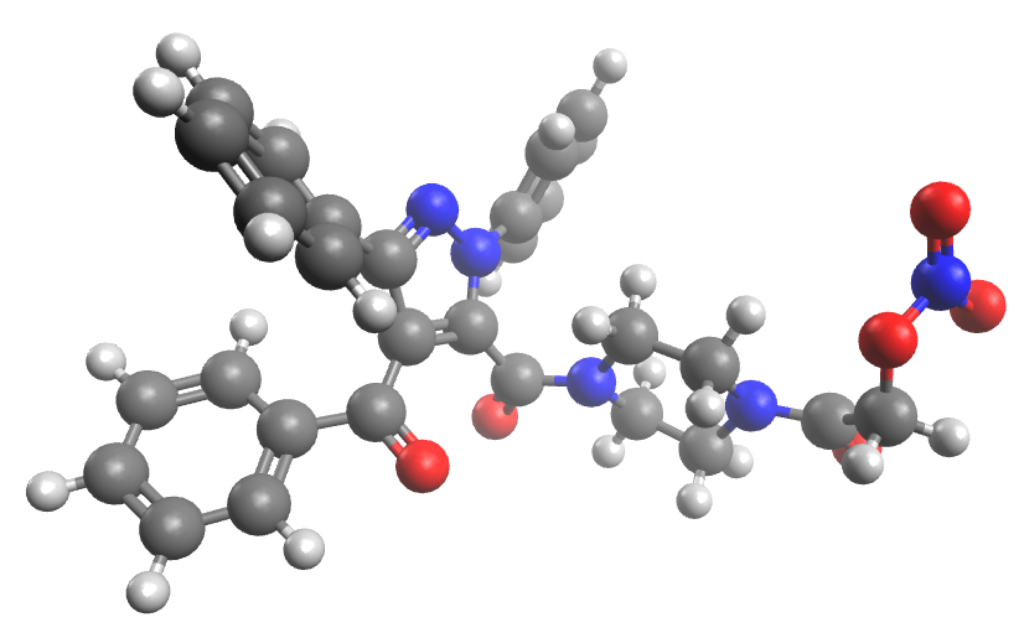
- We aligned all the conformers against the reference lowest energy conformer of the drug molecule and quantified the route mean squared deviation (RMSD, Å) as a measure of ‘likeness’ and compared this to the relative energy of the conformer (kcal/mol). We did this using the universal force field (UFF) and the PM6 semi-empircial method.
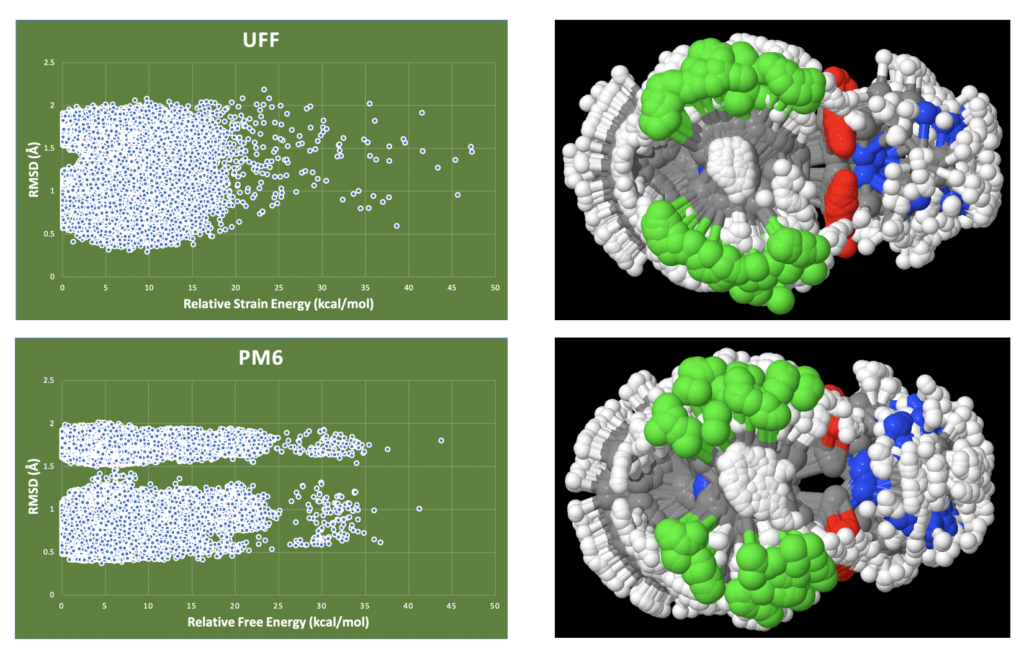
The differences between classical mechanics and quantum are striking. Firstly, the number of conformations at the UFF level is about 400,000 while at the PM6 level it is about 300,000. Using ConstruQt these were obtained in a half a day or so. We have previously discussed spurious conformational states. The difference between PM6 and UFF is these states which are removed at the PM6 level since it better understands the thermostructural landscape
We used obfit to overlay all conformations at PM6 and UFF with the active core moiety to evaluate the RMSD. Here you will find the visually obvious difference in the plots. While UFF represents a kind of blob, the PM6 data shows clear resolution of two bands of RMSD values at the PM6 level compared to UFF (see range of 1-1.3 and then 2.0 and up). This separation can be understood visually looking at the overlay of one of the molecules (selected at random) in the library (see images at right) where the position of the carbonyl (up or down) is highly resolved at PM6 but not so at UFF. Notice the ‘hole’ in the PM6 data, no hole in the UFF data. This points to the fact that “in-betweener” structures (spurious conformers) at UFF tend to collapse to either ‘up’ or ‘down’ at the improved PM6 level.
This basic result should make analysis much easier and more chemically meaningful for drug developers.
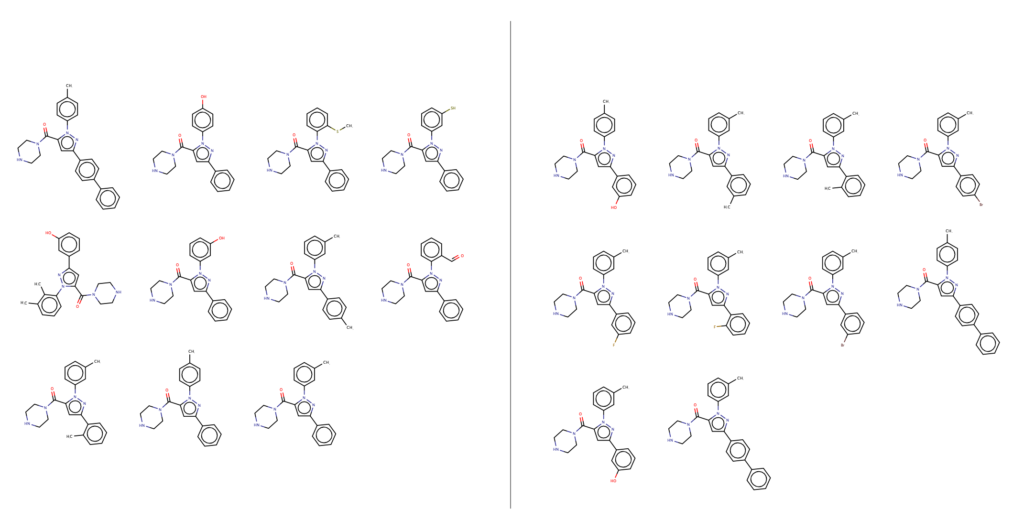
Finally, if you take a look at structures with an RMSD less than 1 and an energy less than 5 kcal/mol (best candidate molecules/conformers) you find that there is only a 30% overlap between UFF and PM6 in terms of which molecules represent that space. In other words, the qualitative result is substantially different. At this point we can not say which is ‘better’ but certainly one should be suspicious of force fields and the quantum chemical results are ‘cleaner.’
The two sets (UFF and PM6) of best representative structures are below.
[…] (2) can we change this outcome, what if we want to have the 1,3-regio product? In a previous post we described a medicinal chemistry study where the 1,3-regio example was, in fact, […]