
InteraQt: Fully Automated Quantum Chemical Supramolecular Chemistry
To the extent that molecular (al)chemistry* has evolved into an applied science the next frontier has become supramolecular chemistry. The recent ‘nods’ to Jean-Marie Lehn (Nobel, 2006) and Frazier Stoddard (Nobel, 2016)** codifies this focus in chemistry going forward. The overarching goal in the study of how molecules interact is to predict and design materials with pre-defined properties. Whether this supramolecular interaction is between a complex bio-polymer (protein) with encoded 3D topology and a small molecule (drug) or whether it involves the periodic interactions that define the unit-cell of a molecular crystal, the study of interactions is confounded by the degrees of freedom involved in how two or more entities may orient with respect to each other in space. Molecules are complex as it is, with configurational spaces defined by rotatable bonds, now add two molecules together and ask, “how might these two complex entities interact with each other?” The complexity is exponential and the resulting ‘material’ properties are subtle.
Chemistry has long used modern digital and mathematical constructs to address molecular and supramolecular complexity. Molecular modeling has been invaluable in molecular chemistry to navigate molecular shape (degrees of freedom) and has been prodigiously applied in medicinal chemistry to design drug-target interactions and streamline the discovery of new therapeutics. Slow to respond to the interaction problem has been the most advanced and accurate ab initio methods in computational alchemical modeling and quantum chemistry due to the limitations of computing power. A recent focus has been on adjusting these quantum chemical methods to better treat the interaction of molecules by applying empirical adjustments through dispersion correction technique lead by Grimme and others. Thus, we have arrived at a place in alchemical digital technology where we can relatively quickly, and very accurately, model molecular interactions using quantum mechanics, yet this approach remains a largely academic pursuit and is not well-applied in industrial research. Imagine a democratized approach to predicting material (or bio) properties using state-of-the-art physics tuned to identifying fundamental properties to connect to emergence phenomena.
Enter InteraQt, ChemAlive’s most recent software development. InteraQt is a fully automated approach to sample 3D interaction space of two molecules and to refine these interactions using modern dispersion-corrected semi-empirical or ab initio quantum mechanics thus allowing the high throughput and accurate prediction of molecular interaction energy and configuration. Piggybacking off the ConstruQt software (see here and here) InteraQt can take any two molecules and automatically sample their supramolecular configurational space using a protocol involving molecular mechanics, molecular dynamics, semi-empirical quantum mechanics and ab initio quantum mechanics. Thus, the input is two SMILES strings, and the output is a set of ‘poses,’ or atomic coordinates, and their associated energy. These pose energies are corrected with thermal harmonic frequency contributions and thus the association pose free-energy is simply the pose energy minus the sum of the energies of the monodispersed molecules.
After the initial optimization at the molecular level, the next step (first InteraQt step) is to sample the configurational space using molecular dynamics. While molecular dynamics is technically best suited to studying time dependent phenomena, it is also incredibly useful as a configurational space sampler, assuming one takes proper precautions to avoid over sampling equilibrium configurations. Thus, InteraQt first starts from 6 starting configurations of the two molecules arranged along their cartesian coordinates and runs 6 independent simulations each lasting 600 picoseconds (Figure 1).
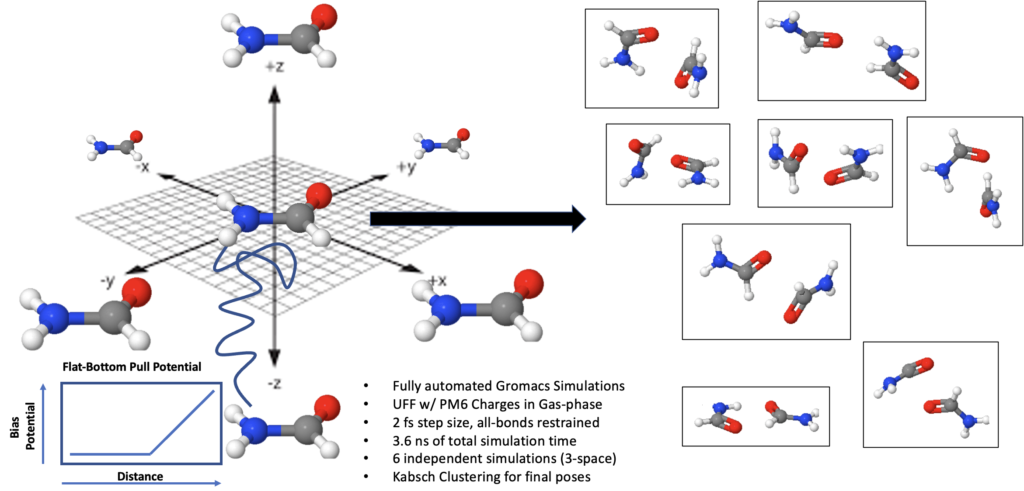
Figure 1. InteraQt sampling approach where 6 simultaneous simulations are run to sample unbiased space and find the most relevant poses.
This is done in order to remove the bias from the simulations. If one had started from a single configuration an ran a longer simulation of 3.6 nanoseconds our test show that the configurational space is poorly sampled and most of the simulation samples a localized equilibrated environment, wasting simulation time and missing key configurations (often even the global minimum region). We have exhaustively tuned and optimized the simulation time and approach. For example, we have found that the application of a ‘pull’ procedure increases the efficiency of the simulation with a flat-bottom potential that keeps the molecules in contact within the solvent box. Simulation in most solvents is possible, but molecular force fields are currently limited to UFF where a topology can be automatically generated suing open-source tools. In order to improve the UFF accuracy we have used semi-empirical PM6 charges instead of standard FF charges. After all six simulations complete their results are aggregated and the Kabsch clustering method is applied to reduce the configurational complexity and return only the most relevant generated poses. These poses are subsequently optimized with semi-empirical PM6-D (dispersion correction). We have tested our protocol against the s22 benchmark set (Figure 2), which comprises 22 basic molecules with very accurate computed dimer energies (e.g. acetic acid, benzene).
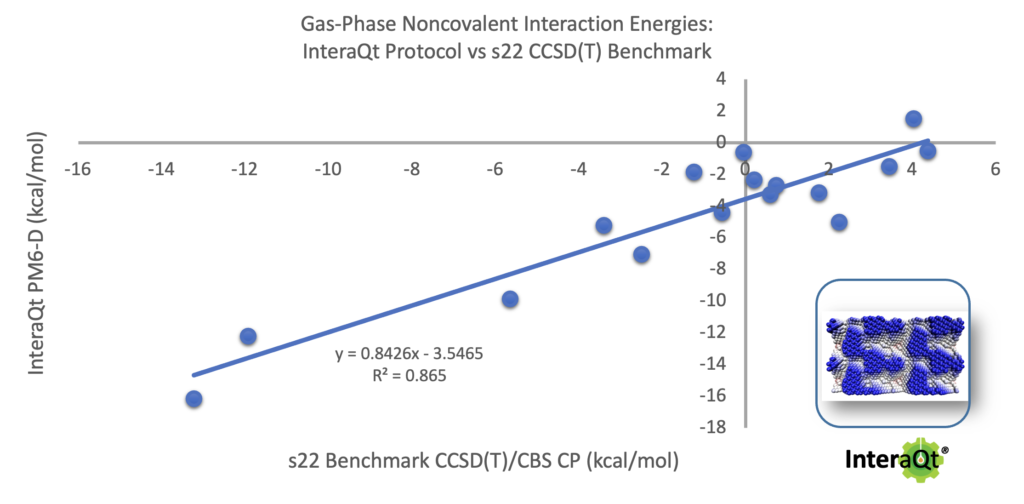
Figure 2. InteraQt protocol results on the s22 Benchmark set.
The tests show a strong correlation to these high-level ab initio association energies (R2 = 0.88) without any further manipulation or refinement indicating that the protocol hits the lowest energy dimer and produces a qualitatively excellent match to much more powerful quantum mechanical approaches.
To demonstrate the utility of this approach further we ran the code on cloud resources for 501 dimers (took about 20 hours) with known experimental flashpoints. The idea is that the flashpoint is related to the vapor pressure, which is an indication of molecular self-stickiness, i.e., the stronger the self-interaction of a molecule the higher the flashpoint because you need to add more heat to achieve the correct vapor density to propagate the combustion process. The results are shown in Figure 3.
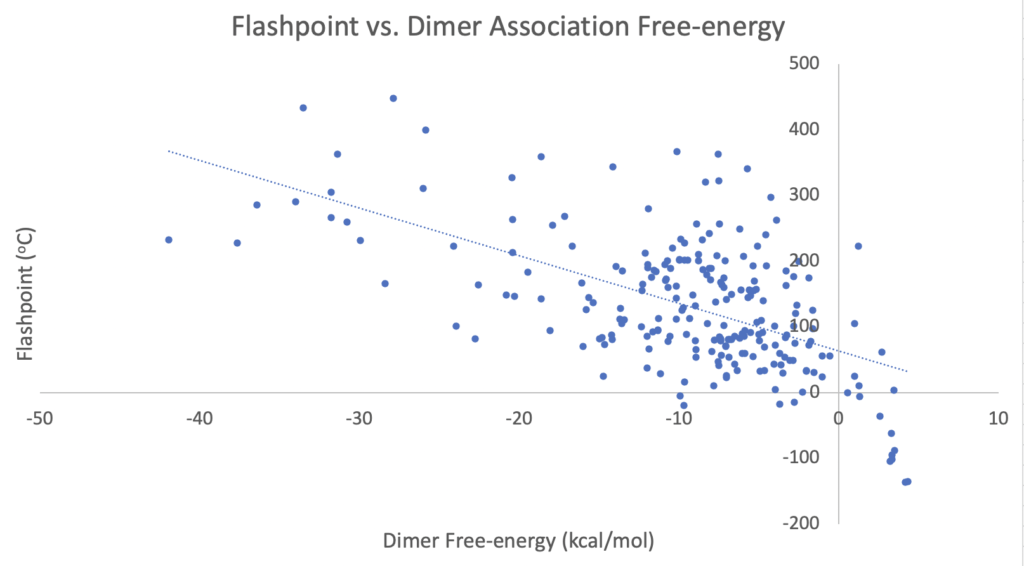
Figure 3. Use-case for InteraQt where we show a correlation between dimer association free energies and experimental flashpoint data.
A reasonable correlation (R2 = 0.33) given the tenuous link and zero methodological tuning.
In conclusion, with InteraQt one can quickly assess molecular interactivity using structural and energetic considerations and establish the first level beyond the molecular in their (al)chemical design. Beyond this complexity is the issue of emergence. Emergent phenomena seemingly arise out of complexity itself and are difficult to connect to the basic unit of construction that forms a material. A crystalline property such as ‘hardness’ does not relate in any obvious way to the molecular structure but emerges out of the crystal structure. However, to the extent that we can connect the molecular length-scale properties of molecules to the nanoscopic length-scale properties of materials and beyond, we should have in place the tools to make predictions to do so taking the step beyong single molecules and looking in detail at molecular dimer interactions. Further work will allow the automatic construction of more complex system involving many bodies to start to really predict materials properties. Get in touch to try it yourself at info@chemalive.com
*In case you are wondering why I use alchemy/alchemistry instead of ‘chemistry.’ Its because German enlightenment figure named Georgius Agricola went around in the 16th century and ‘purged’ the Arabic definitive article from lots of words so he could ‘latinize’ them and pretend like Christian Europe owed nothing to Arab scholarship and history at the onset of the enlightenment. I disagree, its ‘alchemy’. The definitive Arabic article reminds us of the deep historical roots of our craft and mends the bridge between the early phase of research and the modern one. Imagine if early astronomers were instead called ‘astrologers’, that is what we do when we call early chemists ‘alchemists’. Alchemist implies magic / occult*** when in fact early chemical researchers made numerous discoveries and establish ways of thinking that we still use today (they also did cool magic).
**In case you wonder who the hell I am, I have published with Lehn and was taught supramolecular chemistry by Stoddard.
***Khēme means ‘darkness’ in Coptic / Egyptian and refers to the black earth around the Nile River basin (i.e. Egypt), as opposed to the red earth of the dessert. Thus, alchemy translates to the ‘Egyptian Arts’ or even better, the ‘Black Arts’, which sounds very wizardly to me. Its nice to know history.