
Surfing the Potential Energy Surface – Automatic Reaction Analytics with Quantum Chemistry
Experimental synthetic chemistry is a roughly dismal empirical science where the best practitioner cannot expect to be successful more than 1 in 10 attempts. Where an R&D yield of 30% is considered amazing and a fully developed manufacturing process accepts massive waste in atom economy as expected loss (almost 50 billion a year wasted). Most practicing chemist still use textbook knowledge to predict reaction efficacy (“sterics” they say) relying on age-old models developed last century using only a few data points that often have more exceptions to the rules than examples that follow them.
For the last few decades chemists have relied on database driven tools that catalog known reaction outcomes to infer reaction efficacy or repeat a given recipe. They have used Beilstein, Reaxys and Scifinder to this end. Even if you are repeating a reaction recipe, one can expect some difficulty in obtaining the intermediate or product, but when your reaction doesn’t exist in these databases – good luck. Even small changes in composition, shape, electronics, can vastly alter a reaction outcome. Thus, using known reactions that are ‘similar’ to one’s required reaction can be fraught with challenges often requiring weeks of reaction optimization to obtain a reasonable yield. But this is what we do, right?
These days a number of new software solutions have entered the market to help chemists predict which reactions to use to obtain a given desired product. Most of these are based on machine learning technology (Chematica, Chemplanner and IBM Rxn). At their base, they also use massive literature precedent, user-supplied scoring or hard coded rules to forecast the outcome of unknown reactions – they just do all this faster and more efficiently than a chemist pushing buttons on Scifinder.
Of course, it is possible to predict the outcome of reactions without rules based on literature precedent. One must simple use the Schroedinger equation. Quantum chemical reaction prediction has been an effective tool in academia at least since the advent of DFT. The Schroedinger equation is good at computing molecular energy. Molecular energy is the currency of all of molecular chemistry. If you have a good estimate of molecular energy, everything else about its reactivity follows. More recently, advances in solvent modeling and accounting for dispersion and other important effects have rendered quantum chemical reaction prediction a highly accurate and informative tool. However, it is currently too slow, too difficult to set-up and requires expensive (often poorly documented) software and infrastructure to address even the most basic reactions.
Imagine a world where a chemist can take their daily chemical drawings (we call those molecular graphs or line-drawings) and enhance them by automatically generated and accurate molecular reaction energies (delta G rxn) estimates (Scheme 1).
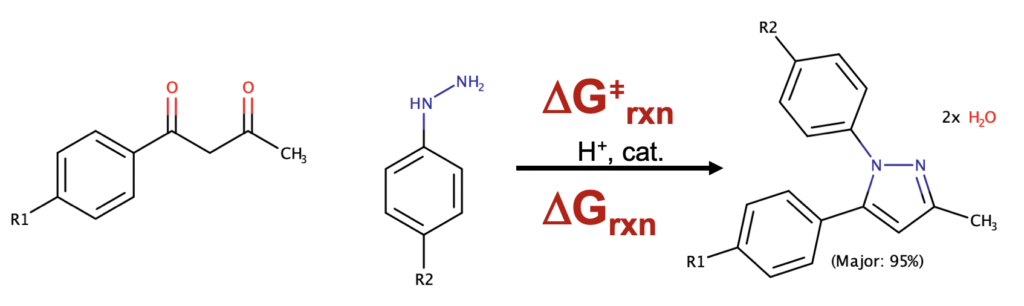
Scheme 1. Main idea: Adding accurate reaction energetics to standard chemical drawings
Further imagine that rates of reaction through estimates of reaction kinetics (delta G double dagger) using transition-state theory are also readily available. Having both the energetics (free-energy) and kinetics (rates) allows the full definition of a reaction route.
ChemAlive has been working towards this end. It has beta tested its software ConstruQt which manages molecular structure and the automatic use of quantum chemical methods to achieve molecular energy estimates quickly and is now piloting its software ReaQt – for automatic reaction analytics. Here is how it works as illustrated by example.
Let’s take the example of the Knorr Pyrazole reaction (Scheme 1). In this reaction a 1,3-dione reacts with a hydrazine to give a pyrazole (oh don’t forget two beautiful molecules of water as well – quantum chemists can’t be sloppy we fear Schroedinger, Bohr, Debroglie, Einstein, Pauling, Mulliken, Huckel etc at the pearly gates). The reaction is known to be regioselective for the 1,5-regio product (as shown) when we are reacting an aryl-1,3-dione and an aryl hydrazine, e.g. we expect that the aryl groups will end up adjacent to each other as configured about the pyrazole core moiety. This regio-selectivity is sensitive to electronic effects, but is generally giving a regiomeric excess of 95% for the 1,5-regio. That’s all fine and good but (1) can we prove it is correct with quantum chemistry (2) can we change this outcome, what if we want to have the 1,3-regio product? In a previous post we described a medicinal chemistry study where the 1,3-regio example was, in fact, desirable.
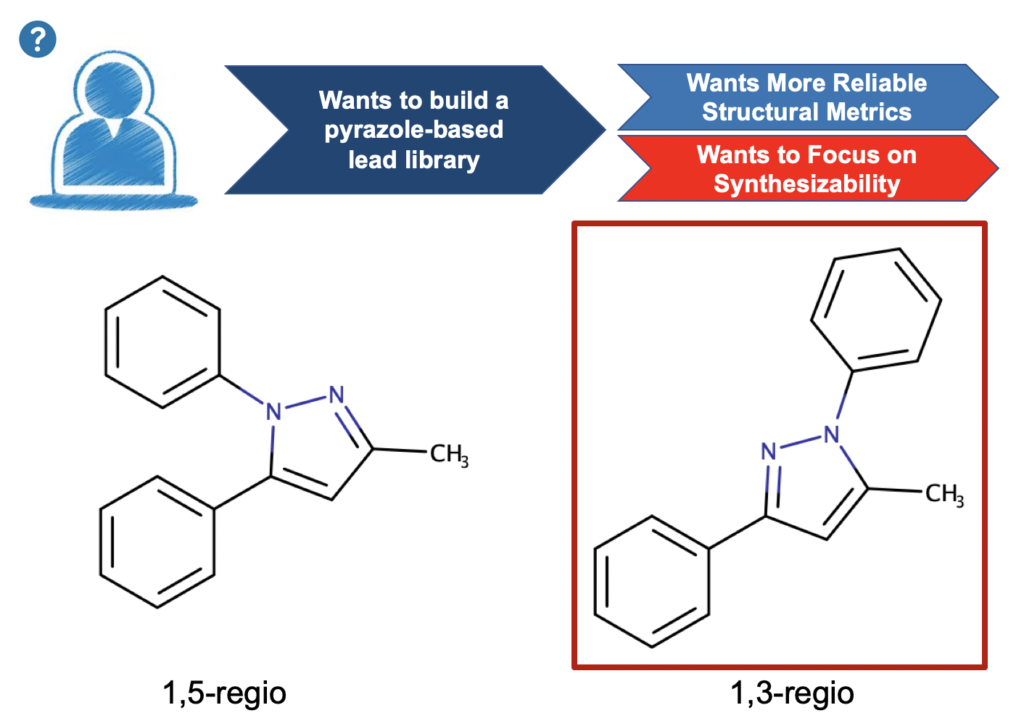
Scheme 2: Previous medicinal chemistry study where our hypothetical med chemists wants the 1,3-regio product for a library study in lead optimization with focus on synthesizability.
Firstly, computing this reaction will require a good understanding of its mechanism. I have proposed one below. In order to address the questions above we will need to calculate at least 15 molecules and 7 transition-states per regioisomer. That’s 44 calculations. (Wouldn’t it just be easier to order these reactants and throw them in a vial, but I digress – this is why quantum chemistry is mainly still an academic pursuit.)
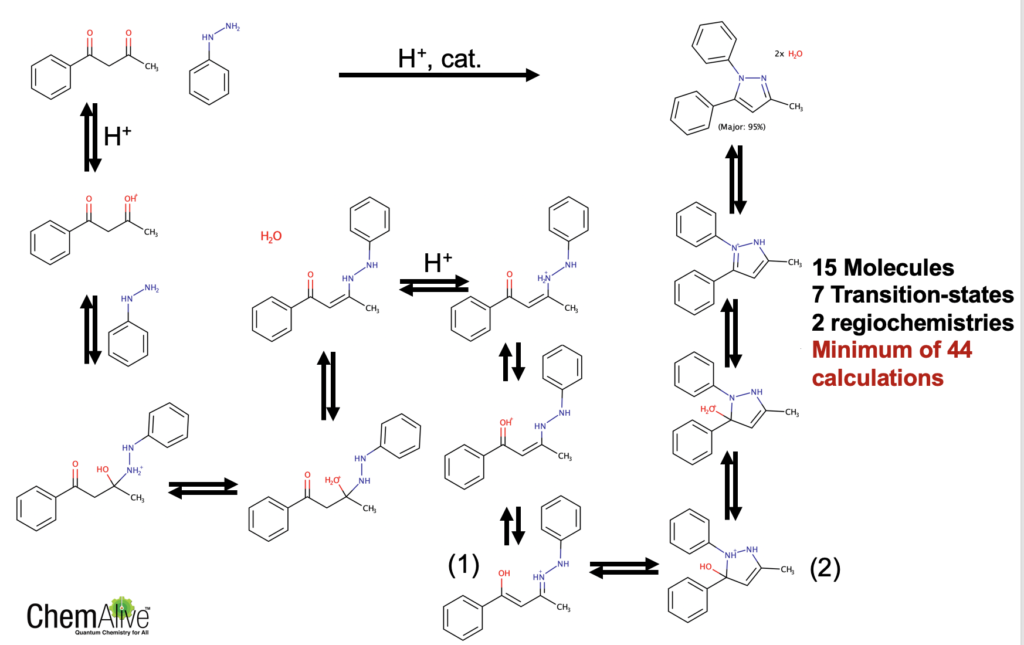
Scheme 3: Proposed mechanism (only 1,5-regio is shown).
To make this blog post shorter, lets assume that the cyclization step is the rate determining step (RDS) from (1) to (2) in Scheme 3 and that we can arbitrarily choose one stereochemical outcome for the immediate intermediate after this barrier (that ammonium group is pseudo-chiral! This gives pseudodiastereomers).
With ConstruQt we can automatically do density functional theory calculations on all 44 molecules directly from their SMILES format. Done. We can then use the ReaQt algorithm to obtain the barrier for the cyclization step directly from the SMILES of the immediate reactant (1) and the SMILES of this immediate product (2). The algorithm (1) Computes the reactant and product in its lowest energy conformation, (2) identifies the atom indices of the atoms involved in the forming bond and their corresponding indices in the reactant and product, (3) scans the bond in suitable increments to generate an initial potential energy surface (PES) then identifies the best guess structure for the transition-state optimization. We should note that we always scan from the product to the reactant for a condensation reaction (reverse for a fragmentation), but since the product is more rigid sometimes we can get some saw-toothing (shark finning, if you like) in the PES. I.e. sometimes our PES is not at equilibrium. To mitigate this we use our algorithm called Reverse-Return (RR) which allows us to scan in both directions to achieve the true minimum energy pathway. Results are here for the 1,3-regio and 1,5-regio product.
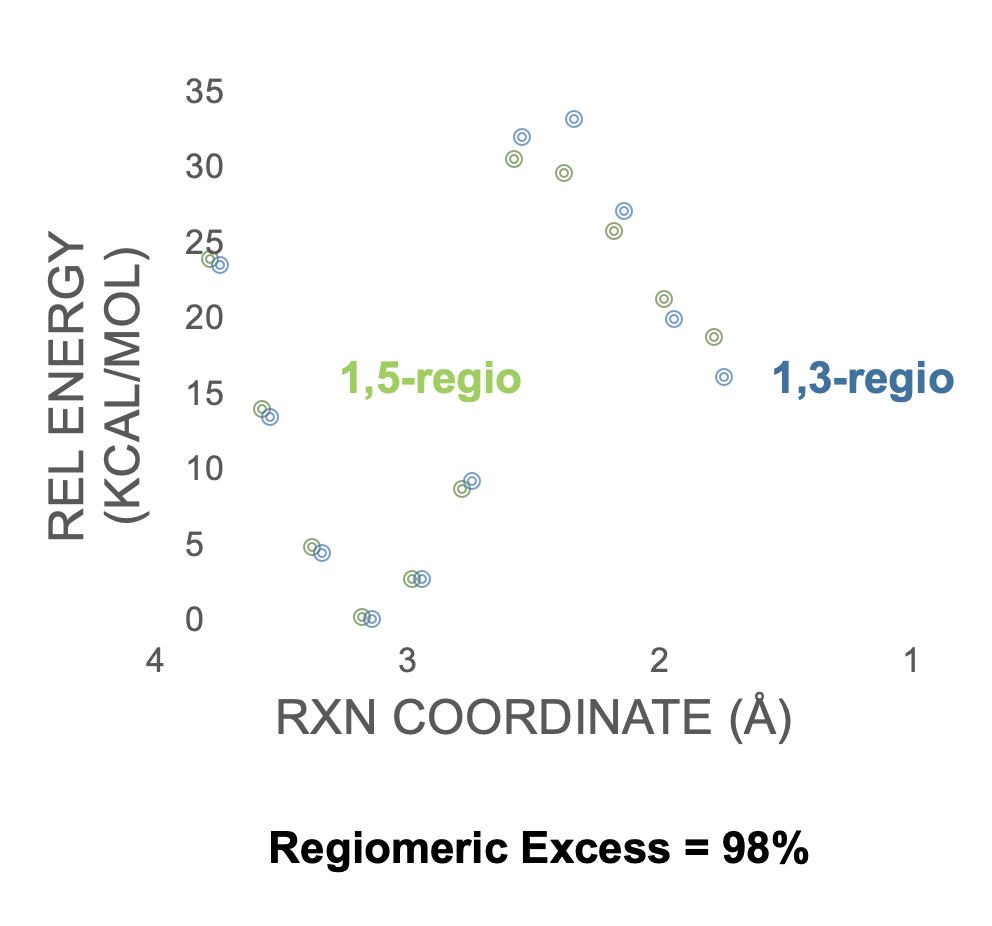
Figure 1: PES for 1,3- and 1,5-regio for the model Knorr Pyrazole example.
If we take these rough barrier heights form the PES and apply the Eyring expression we arrive at a regiomeric excess of 98% for the 1,5-regioisomer in good agreement with expectations.
This analysis, unfortunately took at least 24 hours (more on that later) using B3LYP/6-31+G(d) with COSMO solvation in water. However, it has validated the approach and we can now try to figure out how to change the course of the reaction. Luckily in a previous blog post we discussed amide substituted aryl-1,3-diones and using ConstruQt to build structural libraries with quantum chemistry.
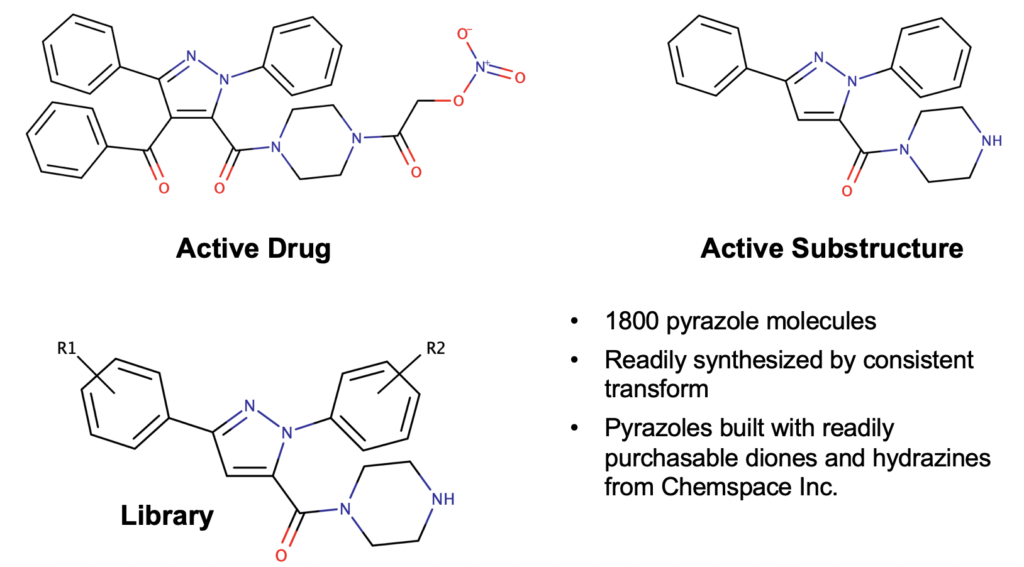
Scheme 4: Previous approach to pyrazole ligand library.
As it turns out that amide is just the thing we need. (To be completely honest, I did not hark this result – just lucky sometimes). Here is the PES for a member of that molecular library towards the 1,3-regioisomer and the 1,5-regioisomer.
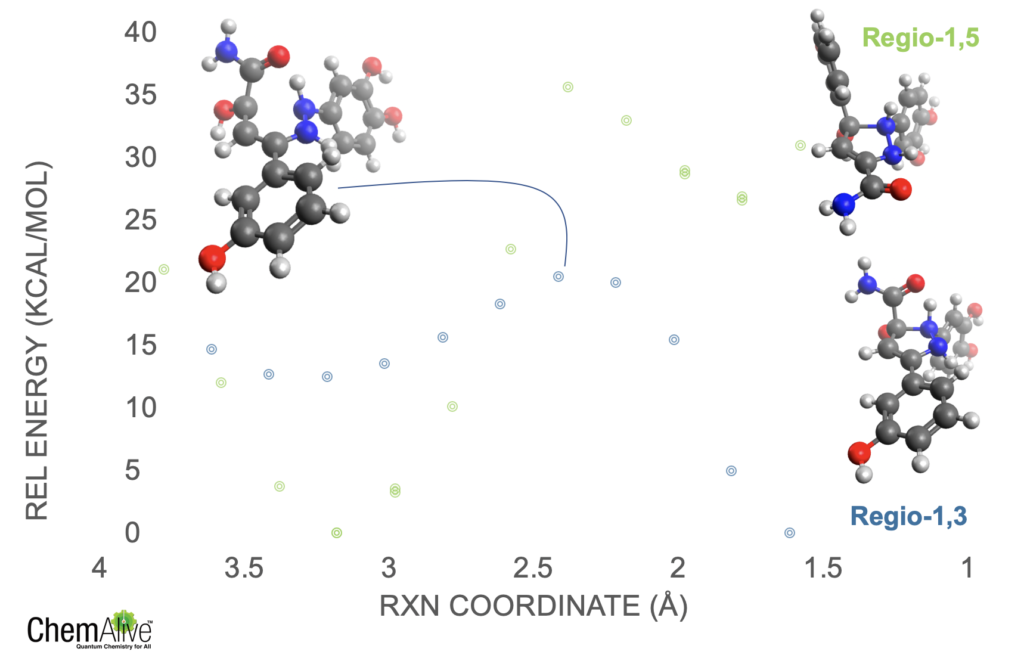
Figure 2: PES for an amide substituted example showing the profound change in relative barrier height from amide hydrogen bonding.
The barriers are significantly different and even the energetics have been altered such that the 1,3-regioisomer is far more favored. Looking at the structures it is clear that this is due to the amide forming a solid hydrogen bond across the reacting bond in the transition-state. This lowers the barrier of this reaction route so significantly that we could expect near perfect regioselectivity for the 1,3-regioisomer.
Now, this PES took a while to generate, but given the fact that one only needs the smiles of the reactant and product on either side of the barrier it took 2 minutes to set-up and provided a key insight that is not readily available from 2D line-drawings, which also provide no energetic insight. ChemAlive continues forward to develop faster methods that will make such insights available in seconds though the application of tuned semi-empirical methods as well as machine learning.
In the meantime, we can help you optimize your reactions. Get in touch info@chemalive.com