
ConstruQt White Paper
ConstruQt – a Reliable Molecular Structure Predictor
Since August Kekulé’s proposal for the tetrahedral configuration of carbon or his more famous realization that benzene was a cyclic molecule, a snake biting its tale, molecular structure has been the leading consideration for the design of new molecules as drugs or performance materials. For the former, it is said that 70% of drug design is based on molecular shape with the remainder attributed to electrostatic or non-bonded interactions.
Structural chemistry began around the 1860 with these dual assignments by Kekulé but it wasn’t until one hundred years later with Allinger’s initial force field approaches that the first classical molecular mechanics (MM) models became available to make computer-assisted prediction of molecular structure. These models themselves are based on principles derived by Robert Hooke, a contemporary of Isaac Newton, in the mid 17th century with additional layers from van der Waals (19th century) etc.
The application of modern physics in the form of the quantum theory / mechanics (QM) developed in the first half of the 20th century by Schroedinger, Dirac and others represents a vast Improvement in scope and accuracy compared to MM. However, persisting to the present, MM approaches to molecular structure and shape prediction dominate across chemistry, despite its age and widely recognized and severe limitations. It survives because it can be applied with unmatched speed allowing it to scale over molecular size (towards proteins and microscale structures) and over library size (up to millions of molecules) whereas quantum mechanics has traditionally been orders of magnitude slower when applied to computational prediction. However, today there is no excuse, except for expediency and tradition, to continue to make predictions with MM with the advent of cloud computing and efficient alternative algorithms and tools to access quantum mechanics such as the faster density functional theory (DFT) and the ultra-fast semi-empirical methods.
ChemAlive (www.chemalive.com) has developed ConstruQt, (https://www.chemalive.com/construqt-api/) to drive the shift away from MM to QM that is coming over the next few years. ConstruQt is high throughput quantum mechanics (QM), deployed on the cloud with full automation and is a first-of-its-kind tool for scaling molecular library design with the power of quantum mechanics. It dramatically increases molecular library accuracy in shape while adding energy-regime predictions for molecular structure such as tautomeric and diastereomeric selection and prioritization – both key to the design of active molecules.
A recent study of 750 conformationally diverse molecules has confirmed the extent to which classical force field approaches can be unreliable compared to higher-level data. The study further demonstrated the utility and accuracy of fast semi-empirical quantum mechanics as a paradigm shifting method in structural analysis. Thus, if molecular shape is being used in a molecular design workflow it is certainly generating false positives or filtering-off potential leads that would be captured by quantum mechanics. In addition, QM adds increasingly important scope towards reaction prediction and spectroscopic assignment.
Independently, we at ChemAlive have analyzed over 3 million molecules and over 200 million conformations using the Universal (UFF) and Merck Molecular (MMFF) force fields compared to the semi-empirical QM method PM6.
Our analysis reveals important differences in the global treatment of molecular shape by these methods. First of all, force fields over estimate conformer energy for strained systems thus creating false negatives and throwing out accessible conformers. The distribution in Figure 1 (a) shows a long tail for the UFF method reaching out to 20 kcal/mol.
(a) (b)
(b)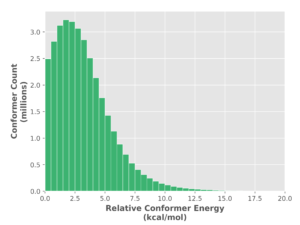
Figure 1. Histogram of optimized relative conformer energy (kcal/mol) at (a) universal force field (UFF) and (b) PM6 for 68 million and 35 million conformers, respectively.
While the majority (over 90%) of structures are within 5 kcal/mol of the minima, most molecules have at least a few strained shapes. The PM6 distribution (Figure 1 (b)) is tighter extending to 12.5 kcal/mol, indicating that UFF produces a significant number of false outliers. Most importantly though, UFF produces numerous false structures (spurious minima) as there are 2 times less PM6 structures than UFF structures (Figure 1 (a) vs. (b)). All UFF structures are submitted for PM6 calculation, thus the structures which were considered divergent (according to an RMSD analysis) become convergent at PM6. This results in considerably less conformations predicted at the semi-empirical level, reducing the scale of conformational analysis and thus its complexity.
ConstruQt provides improved accuracy as evident in the error analysis (Figure 2).
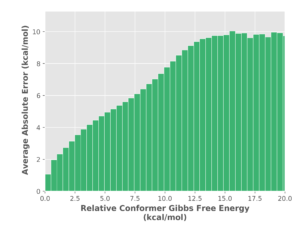
Figure 2. UFF versus PM6 relative conformer energy difference (error) as a function of absolute conformer relative energy (PM6).
The error in mechanics force fields is 90% to 200% of the relative energy predicted by PM6 – false positives and negatives are pervasive as the molecules adopt more diverse structures and thus the error is correlated to absolute relative energy. To further validate our conformational searching method and energetic accuracy, ChemAlive processed 47,000 organic molecular crystals from the Crystallographic Open Database (http://www.crystallography.net/cod/). Our current algorithm was able to predict the XRD structure as a conformer most of the time. For these cases, the XRD structure was within 5 kcal/mol of the predicted global minimum. Further improvement are being applied to increase the predictive power.
In addition to shape, ConstruQt automatically manages key tautomeric forms. Of the 1.5 million substances in our database, 1.8 million are tautomers. Thus, half of all substances have at least one other tautomeric form that is relevant for investigation. On the other hand, many tautomeric forms that might be considered are energetically inaccessible (Figure 3) and can be readily discounted in library analysis, even after considering specific binding motifs in protein-ligand interactions, for example.
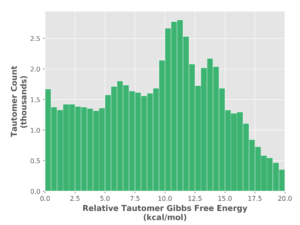
Figure 3. Analysis of 0.6 million tautomers showing the tautomeric relative Gibbs free energy.
This is only evident from a quantum mechanical energetic analysis and would not be available from a classical mechanics approach. The noteworthy structure in the Figure 3 stems from the limited types of tautomeric arrangement considered of biological import and perhaps from the statistical power of the smaller dataset.
ConstruQt can process up to 500 molecules per minute per server matching your current high throughput requirements and enhancing the utility in screening applications. It can scale to thousands of CPUs in minutes and uses inexpensive spot computing. ConstruQt will deploy calculations of your molecules on an AWS spark cluster in minutes based only on SMILES list input and return quantum chemical data in an SD file ready for integration with standard cheminformatics and drug discovery platforms.
Please try it here by simply filling out the form with a list of SMILES of your choosing (up to 25 for testing):
https://www.chemalive.com/construqt-api/
For larger libraries and advanced quantum mechanics methods for increased accuracy or advanced properties (spectra, charges, reactions), please get in touch at info@chemalive.com.